Sección 7 Validación del modelo QSAR
Un buen modelo debe cumplir con varios criterios en términos de predictibilidad y rendimiento para ser aceptado. Estos criterios de evaluación se categorizan generalmente en validaciones internas y externas.
Metodos de validacion
Después de generar modelos QSAR, normalmente se realiza el procedimiento de validación interna para seleccionar los modelos QSAR iniciales. Una vez que se cumplen los criterios requeridos, conocidos como validación interna, la capacidad predictiva de los modelos candidatos seleccionados se evalúa mediante el procedimiento de validación externa.
Validación interna
Se utilizan los mismos datos (compuestos moleculares) que se usaron como conjunto de entrenamiento. Todos los criterios utilizados con este propósito son simplemente indicadores de la calidad de la evaluación interna de los modelos propuestos. Sin embargo, la capacidad predictiva del modelo puede no generalizarse para los nuevos compuestos.
Validación cruzada interna: La validación cruzada interna implica excluir un pequeño subconjunto de compuestos y aplicar el modelo utilizando el resto de los datos para predecir la actividad biológica de los compuestos excluidos, simulando un conjunto de pruebas. Esto se repite hasta que todos los compuestos se hayan considerado como el subconjunto excluido y se utilizan para predecir valores de actividad biológica.
Validación cruzada dejando uno fuera: También conocido como LOO (leave-one-out), este método es comúnmente utilizado para la evaluación inicial de un modelo QSAR. En este enfoque, se excluye un compuesto del conjunto de datos completo y se entrena el modelo utilizando el resto de las moléculas. Luego, se utiliza el modelo entrenado para predecir la actividad del compuesto excluido. En este proceso, cada compuesto se excluye uno por uno hasta que se hayan predicho todos los valores de actividad al menos una vez. El coeficiente \(Q²\) se emplea para determinar la capacidad predictiva del modelo en este método y se calcula de la siguiente manera: \[Q^2=1-\frac{\sum(Y_{{{obs}}({train})}-Y_{{{pred}}({train})})^2}{\sum(Y_{{{obs}}({train})}-\bar{Y}_{({train})})^2}\] Donde \(Y_{{obs}}\) y \(Y_{{pred}}\) son los valores observados y predichos de actividad, respectivamente. Y \(\bar{Y}\) se refiere al promedio de los valores de actividad. El valor mínimo aceptable para \(Q^2\) es 0.5, y es evidente a partir de la ecuación que su valor varía de cero a uno. El error de predicción de la desviación estándar (SDEP) se calcula mediante la siguiente ecuación: \[SDEP = \sqrt{\frac{\sum(Y_{{{obs}}({train})}-Y_{{{pred}}({train})})^2}{n}}\] Donde n indica el número de puntos de datos (compuestos moleculares)
Validación cruzada de exclusión de grupo: En este método, en lugar de excluir sólo un compuesto a la vez, se excluye un número definido de compuestos cada vez, En este método, el conjunto de datos completo se divide en k grupos, y en cada repetición del proceso, se predicen las actividades biológicas del grupo excluido utilizando el modelo entrenado basado en los compuestos restantes. El valor correspondiente de \(Q²\) también se determina de manera similar al método de validación cruzada dejando uno fuera.
Parámetro \(r_{m}^2\): Un alto valor en \(Q²\) no necesariamente refleja la buena capacidad predictiva del modelo. En algunas ocasiones, su elevación podría surgir debido a diferencias amplificadas en el rango de valores observados en comparación con la respuesta media (es decir, \(\sum(Y_{obs(train)}-\bar{Y}_{(train)})²\) por lo tanto, en la ecuación, a medida que esta diferencia crece, también lo hace el valor de \(Q²\). Esto se relaciona con situaciones donde los valores observados (los valores reales) se encuentran dispersos en comparación con la respuesta promedio. Esto significa que los valores reales pueden variar sustancialmente alrededor de la media. Cuando la dispersión entre los valores reales es considerable, indica una notable variabilidad en los datos. Esto podría ocurrir, por ejemplo, cuando los valores reales están distribuidos a lo largo de un amplio rango de valores sin seguir un patrón claro. El punto central es el siguiente: en casos donde los valores reales se dispersan ampliamente, el índice de calidad \(Q²\) puede incrementar incluso si el modelo no es especialmente competente en predecir esos valores dispersos. Esto se debe a que Q considera la variabilidad global de los datos y compara la actuación del modelo en relación con esa variabilidad. Si los valores reales presentan mucha variación, el modelo podría lograr un alto valor en \(Q²\) al capturar simplemente parte de esa variabilidad, aun si no realiza predicciones precisas. En otras palabras, el modelo podría reflejar la tendencia general de dispersión en los datos, sin necesariamente predecir con precisión los valores individuales. Este escenario podría llevar a una apreciación exagerada del rendimiento del modelo cuando se emplea \(Q²\) como métrica de evaluación, ya que \(Q²\) tiene en cuenta la variabilidad total de los datos. Para resolver este problema, se implementó el parámetro \(r_{m}^2\). Las ecuaciones para calcular dicha métrica se muestran a continuación: \[\bar{r²_m}=\frac{(r²_m+{r'}²_m)}{2}\] \[\Delta r²_m=|r²_m-r'²_m|\] \[r²_m=r²×(1-\sqrt{r²-r²_0}), r'²_m=r²×(1-\sqrt{r²-r'²_0})\] \[r²_0=1-\frac{\sum(Y_{obs}-k×Y_{pred})²}{\sum(Y_{obs}-\bar{Y}_{obs})²}\] \[r'²_0=1-\frac{\sum(Y_{pred}-k'×Y_{obs})²}{\sum(Y_{pred}-\bar{Y}_{pred})²}\] \[k=\frac{\sum(Y_{obs}×Y_{pred})}{\sum(Y_{pred})²}\] \[k'=\frac{\sum(Y_{obs}×Y_{pred})}{\sum(Y_{obs})²}\]
En las ecuaciones mencionadas, el término “r²” se refiere al coeficiente de correlación al cuadrado entre los valores que se observan en los datos y los valores que el modelo predice. Esto se hace teniendo en cuenta la intercepción de la línea de regresión. Los valores \(r²_{0}\) y \(r'²_{0}\) prima indican el mismo concepto, pero obligando a que la línea de regresión pase por el origen. Para el término con prima (es decir, \(r'²_{0}\)), los ejes \(X\) y \(Y\) están invertidos. Las pendientes de estos coeficientes de correlación al cuadrado son \(k\) y \(k'\), respectivamente. Los valores recomendados para \(r²_{m}\) (es decir, el promedio de \(r²_{m}\) y \(r'²_{m}\)) y \(\Delta r²_{m}\) (diferencia absoluta entre \(r²_{m}\) y \(r'²_{m}\) deberían ser mayores a 0.5 y menores a 0.2, respectivamente.
Y-scrambling: La aleatorización es un método valioso para evaluar la robustez de un modelo QSAR, es decir, su capacidad para mantener la precisión de sus predicciones en diversas circunstancias y con diferentes conjuntos de datos. Este enfoque implica construir el modelo después de mezclar al azar los valores del punto final (es decir, respuestas biológicas). Se calcula un parámetro llamado \(^c{R²_p}\) se define en base a los coeficientes de correlación obtenidos de los modelos desarrollados antes y después del mezclado (es decir, \(R²\) y \(R²_r\), qué son los coeficientes de determinación y coeficiente de correlación después de la aleatorización, respectivamente). \[^c{R²_p}=R\sqrt{R²-R²_r}\] Si \(^cR²_p\) es mayor que 0.5, esto indica que el modelo original no se basa en coincidencias aleatorias. En contraste, los modelos aleatorizados deberían tener un valor \(Q²\) más bajo. Un \(^cR²_p\) superior a 0.5 sugiere que el modelo puede predecir con precisión incluso cuando se introducen cambios aleatorios en los datos de entrada. En resumen, este enfoque ayuda a probar la solidez del modelo QSAR y su capacidad para hacer predicciones consistentes más allá de los datos originales.
Bootstrapping: El objetivo del bootstrapping es generalizar la relación dentro del modelo desarrollado. En este tipo de método de validación, el conjunto de datos original se divide al azar en conjuntos de entrenamiento y prueba varias veces. Luego, el conjunto de entrenamiento se utiliza de manera similar al método LOO, con la diferencia de que la exclusión de los compuestos se realiza al azar, y un compuesto puede ser excluido una vez, varias veces o nunca. La división del conjunto de datos original en conjuntos de entrenamiento y prueba se realiza varias veces, y el procedimiento de bootstrap se lleva a cabo como se describe anteriormente. Los valores promediados obtenidos de \(Q²_{bootstrap}\) y \(R²_{bootstrap}\) deben cumplir los valores mínimos aceptables para \(Q²\) y \(R²\) obtenidos de la evaluación LOO y deben oscilar cerca de los valores de \(Q^2\) y \(R^2\) para las estadísticas LOO convencionales.
Validación externa
Los compuestos del conjunto de prueba no se utilizan en el entrenamiento del modelo QSAR y, por lo tanto, se emplean en el procedimiento de validación externa.
La selección de un subconjunto del conjunto de datos como conjunto de prueba es el punto de partida para la validación externa. En este grupo, se hacen predicciones sobre las actividades biológicas de los compuestos para medir cuán bien funciona el modelo en términos de predicciones precisas. Un número comúnmente usado para evaluar qué tan bien predice el modelo es el coeficiente de correlación al cuadrado (\(R^2\)), que se calcula usando la ecuación: \[R²=1-\frac{\sum(Y_{obs(test)}-Y_{pred(test)})²}{\sum(Y_{obs(test)}-\bar{Y}_{train})²}\] Donde \(Y_{obs(test)}\), \(Y_{pred(test)}\) y \(\bar{Y}_{train}\) son los valores observados, predichos y promedios de las respuestas biológicas para los conjuntos de prueba y entrenamiento, respectivamente. El valor de \(R²\) varía de 0 a 1, y se sugiere que no debería ser menor que 0.6.
Generalmente se requieren parámetros adicionales para una evaluación más completa, porque el uso exclusivo de \(R²\) no es suficiente para determinar externamente la validez de los modelos QSAR. Algunos de estos criterios, ampliamente empleados para mostrar las diferencias entre los valores observados y los predichos en las actividades del conjunto de pruebas, fueron propuestos por Golbraikh y Tropsha en 2002. En estos criterios, se calculan dos coeficientes de correlación al cuadrado: \(R²_0\) y \(R'²_0\). Estos coeficientes miden la correlación entre las actividades observadas y predichas, excluyendo el intercepto; sin embargo, el último parámetro se calcula considerando ejes invertidos. Las pendientes de las líneas de regresión son \(k\) y \(k'\), respectivamente. Los valores umbral sugeridos para estos criterios se definen como: \[\frac{R²-R²_0}{R²}<0.1 y 0.9 \leq k \leq 1.1\] \[\frac{R²-R'²_0}{R²}<0.1 y 0.9 \leq k' \leq 1.1\] \[|R²-R'²_0| < 0.3\] Las discrepancias entre los valores de actividad biológica observados y predichos del conjunto de prueba en términos de capacidad predictiva externa pueden calcularse mediante la siguiente ecuación conocida como error cuadrático medio de predicción: \[RMSEP=\sqrt{\frac{ \sum_{i=1}^{^nEXT}(Y_{obs(test)}-Y_{pred(test)})² }{^nEXT}}\] Este parámetro no es aplicable para la comparación de modelos con diferentes valores de punto final (actividad biológica) debido a su dependencia del rango de estos valores de actividad. Esto es porque su eficacia depende del rango de valores de actividad que se estén considerando. En otras palabras, si los modelos están evaluando diferentes tipos de respuestas biológicas, este parámetro no sería útil para hacer una comparación precisa entre ellos. Otro criterio similar para determinar la capacidad predictiva externa de un modelo QSAR es el promedio de errores absolutos expresados como: \[MAE=\frac{\sum_{i=1}^{^nEXT}|(Y_{obs(test)}-Y_{pred(test)})|}{^nEXT}\] Cuanto menores sean los valores de RMSEP y MAE, mejor será la capacidad predictiva del modelo desarrollado. Además de los parámetros utilizados para validar externamente los modelos, otros criterios también han sido sugeridos por los modeladores de QSAR. Estos se presentan como funciones \(Q^2\) y se definen en base al valor de \(Q^2\) obtenido en la validación cruzada interna LOO. La primera función Q2 se denota como \(Q²_{F1}\), la cual se calcula utilizando la siguiente fórmula: \[Q²_{F1}=1-{\frac{\sum_{i=1}^{^nEXT}(Y_{obs(test)}-Y_{pred(test)})²}{\sum_{i=1}^{^nEXT}(Y_{obs(test)}-\bar{Y}_{train})²}}\] \(Q²_{F2}\), la segunda función \(Q^2\), tiene una ligera diferencia en el denominador de \(Q²_{F2}\), de tal manera que se calcula la media de los valores observados en el conjunto extremo. Esto puede considerarse una ventaja en condiciones donde no se dispone de información del conjunto de entrenamiento, de la siguiente manera: \[Q²_{F2}=1-{\frac{\sum_{i=1}^{^nEXT}(Y_{obs(test)}-Y_{pred(test)})²}{\sum_{i=1}^{^nEXT}(Y_{obs(test)}-\bar{Y}_{test})²}}\] Aunque tanto \(Q²_{F1}\) y \(Q²_{F2}\) son utilizados por profesionales de QSAR, estos parámetros sufren de una distribución no uniforme del rango del conjunto de entrenamiento, es decir, estos parámetros pueden ser afectados negativamente si los valores de actividad biológica en el conjunto de entrenamiento están concentrados en un rango limitado o desigual. Esto puede llevar a que los parámetros proporcionen medidas de rendimiento inexactas o poco confiables, ya que el modelo puede no generalizar bien a datos nuevos si no se refleja una amplia variedad de valores de actividad en el conjunto de entrenamiento. Esta diferencia ha sido resuelta mediante el desarrollo de un nuevo término denominado \(Q²_{F3}\): \[Q²_{F3}=1-{\frac{\sum_{i=1}^{^nEXT}(Y_{obs(test)}-Y_{pred(test)})²/^nEXT}{\sum_{i=1}^{^ntrain}(Y_{obs(train)}-\bar{Y}_{train})²/^ntrain}}\] Este parámetro no depende de la distribución ni del tamaño del conjunto de datos externo. Sin embargo, su valor se ve fuertemente afectado por la selección del conjunto de entrenamiento.
El parámetro \(r²_m\) utilizado en la validación interna también puede aplicarse de manera similar en la validación externa en los valores de punto final observados y predichos para el conjunto de prueba.
También hay un parámetro conocido como coeficiente de correlación de concordancia (CCC). Este parámetro se utiliza para determinar el acuerdo entre los valores observados y predichos de los puntos finales. En otras palabras, determina a cuán cerca están los valores que se han predicho utilizando un modelo en comparación con los valores reales que se observaron en la realidad. La precisión y exactitud de los modelos propuestos se miden simultáneamente mediante este parámetro. La precisión trata sobre la distancia de los valores observados respecto a la línea ajustada, mientras que la exactitud detecta la desviación de la línea de regresión que pasa por el origen. \[CCC=\frac{2\sum_{i=1}^{n}(Y_{obs(test)}-\bar{Y}_{obs(test)})(Y_{pred(test)}-\bar{Y}_{pred(test)})}{\sum_{i=1}^{n}(Y_{obs(test)}-\bar{Y}_{obs(test)})²+\sum_{i=1}^{n}(Y_{pred(test)}-\bar{Y}_{pred(test)})²+n(Y_{pred(test)}-\bar{Y}_{pred(test)})}\] El valor ideal para el CCC está cerca de la unidad; sin embargo, el valor umbral aceptable es mayor a 0.85. Dicho umbral no es demasiado estricto, sin embargo, cuanto menos dispersos estén los datos (valores observados vs. valores predichos para el conjunto de prueba), más preciso será el modelo.
Todos los parámetros mencionados anteriormente son útiles para evaluar el poder predictivo de los modelos; sin embargo, tienen sus propias ventajas y desventajas. Se recomienda juzgar la predictibilidad del modelo en base a las estadísticas \(Q²_{F3}\) y CCC, que son independientes de la distribución del conjunto de predicción y proporcionan una evaluación más realista sobre la calidad del modelo. En contraste, no es recomendable depender en gran medida de los parámetros \(r²_m\), \(Q²_{F1}\) y \(Q²_{F2}\), que no son estables y tienden a aumentar a medida que aumenta el tamaño del conjunto de datos de predicción.
Dominio de aplicabilidad
El objetivo de un análisis QSAR es predecir cómo responderán las nuevas sustancias químicas. En este contexto, se debe determinar el dominio de aplicabilidad (AD) del conjunto de entrenamiento. AD se refiere al espacio físico-químico, estructural o biológico, conocimiento o información en el que se ha desarrollado el conjunto de entrenamiento, y para el cual es aplicable para hacer predicciones sobre nuevos compuestos. Para definir el AD de un modelo QSAR, se deben utilizar los parámetros más relevantes, generalmente los que son descriptores del modelo. La idea es que el QSAR se utilice idealmente solo para hacer predicciones dentro de este rango específico de condiciones, evitando hacer extrapolaciones fuera de ese rango.
Para determinar el AD es importante la división del conjunto de datos completo en conjuntos de entrenamiento y prueba. Esta división es crucial para definir el dominio de aplicabilidad, que es el rango de condiciones en el cual el modelo es válido. Y la distribución de las moléculas en ambos debe abarcar todo el espacio en términos de estructura química y respuesta, es decir, las moléculas en ambos conjuntos de datos (entrenamiento y prueba) deben ser representativas y variadas en términos de su estructura química y cómo responden en los experimentos. Esto asegura que el modelo esté preparado para predecir bien para una amplia gama de compuestos. El concepto de AD es asegurarse de que el modelo QSAR se aplique sólo a compuestos que son similares a los que se usaron para entrenarlo. Esto significa que el modelo no debería incluir compuestos muy diferentes, ya que podría llevar a predicciones poco confiables para compuestos nuevos fuera de ese rango de similitud. La idea es que el modelo QSAR funcione mejor cuando se entrena y se prueba con una variedad representativa de compuestos, y que solo se utiliza para predecir para compuestos que son similares a los del conjunto de entrenamiento para evitar resultados poco confiables.
Para determinar el dominio de aplicabilidad (AD) en los modelos QSAR, se han desarrollado diversas metodologías para estimar las regiones de interpolación en un espacio multivariado que abarca descriptores y valores de respuesta. Los métodos comunes son: (1) aquellos basados en rangos en el espacio de descriptores, o (2) en el rango de la variable de respuesta, (3) métodos geométricos, (4) métodos basados en distancias y (5) distribución de densidad de probabilidad.
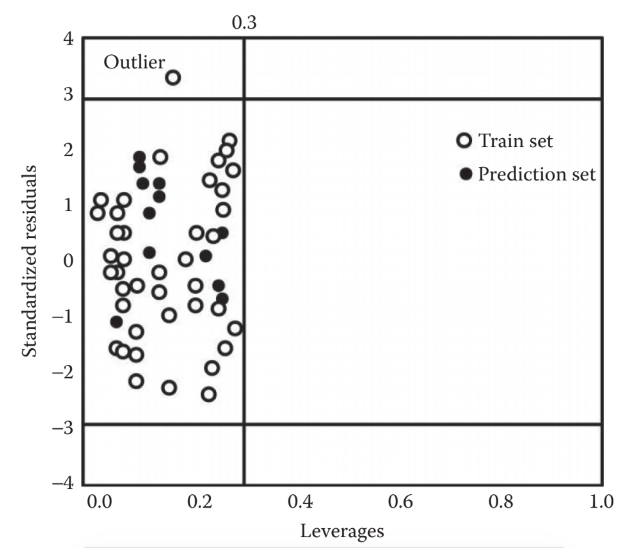
La Gráfica 1 muestra un ejemplo ilustrativo del AD. En ella se comparan los residuos estandarizados (las diferencias entre los valores predichos y observados) con los valores de influencia de un modelo QSAR. Estos valores de influencia indican la distancia a la que se encuentra cada compuesto del centroide (punto central) en un espacio de descriptores. Básicamente, representan la distancia entre el compuesto y el centro del grupo de compuestos similares en términos de descriptores. Para identificar los valores atípicos, se traza un área cuadrada alrededor de los puntos en la gráfica, y se consideran valores atípicos aquellos que se encuentran fuera de esta área, que se define en un rango de ± 3 desviaciones estándar, con un umbral de influencia de 0.3. Estos umbrales son valores arbitrarios para una gráfica de AD de Williams, como se muestra en la Gráfica 1.
Software y herramientas
Las metodologías mencionadas anteriormente están disponibles en programas especializados o generales, ya sea de forma gratuita o comercial. Uno de los programas utilizados para el análisis y la validación QSAR es QSARINS. En él, se implementan todos los pasos necesarios para un análisis QSAR, incluido el análisis de datos, la selección de descriptores, el análisis y la validación del modelo, así como la selección del modelo. Ofrece métodos de validación como la cruzada interna y evaluaciones externas como RMSE, \(Q²_{F1}\), \(Q²_{F2}\), \(Q²_{F3}\), \(r²_m\), \(\Delta{r²_m}\), CCC y los criterios de Golbraikh y Tropsha.
Se ha desarrollado un conjunto de herramientas de validación QSAR para las validaciones internas y externas, así como para el análisis del AD. Estas herramientas se han presentado en diferentes programas y están disponibles públicamente en http://dtclab.webs.com/software-tools. AutoQSAR es un ejemplo comercial que incluye aspectos del análisis QSAR, como análisis de dominio de aplicabilidad y criterios de validación. Estos son solo ejemplos, ya que hay más herramientas disponibles en la literatura.
Desafíos
Se ha desarrollado un conjunto de herramientas de validación QSAR para las validaciones internas y externas, así como para el análisis del AD. Estas herramientas se han presentado en diferentes programas y están disponibles públicamente en http://dtclab.webs.com/software-tools. AutoQSAR es un ejemplo comercial que incluye aspectos del análisis QSAR, como análisis de dominio de aplicabilidad y criterios de validación. Estos son solo ejemplos, ya que hay más herramientas disponibles en la literatura.
Se han establecido reglas importantes para desarrollar modelos QSAR que puedan predecir propiedades químicas. Estas reglas, propuestas por expertos en QSAR en 2002 y adoptadas por la Organización para la Cooperación y el Desarrollo Económico (OCDE) en 2004, incluyen cinco principios clave:
- Punto final claro.
- Algoritmo inequívoco.
- Dominio de aplicabilidad definido.
- Medidas adecuadas de calidad, robustez y predictibilidad.
- Interpretación mecanicista (si es posible).
Estos principios son esenciales para el desarrollo de modelos QSAR confiables con fines regulatorios.
La falta de cumplimiento de las directrices de la OECD por parte de muchos modeladores resulta en modelos QSAR que no son válidos para predecir actividades biológicas. La principal limitación radica en la escasez de datos disponibles para el análisis, y el uso de conjuntos de datos pequeños dificulta la validación externa. La división de datos en conjuntos de entrenamiento y prueba presenta desafíos, y la selección supervisada de datos no se recomienda en ciertos casos debido a su potencial para sesgar el modelo y generar resultados engañosos. Por lo tanto, se insta a la precaución al utilizar este enfoque, y se subraya la importancia de emplear conjuntos de datos representativos y que reflejen adecuadamente la diversidad de las propiedades o respuestas que se intentan predecir. La validación es crucial, y no seguirla puede llevar a modelos poco fiables. En resumen, se destaca la importancia de cumplir con los criterios de validación para garantizar la fiabilidad de un modelo QSAR.